-
Halide Free M(BH4)2 (M = Sr, Ba, and Eu) Synthesis, Structure, and Decomposition

M. Sharma, E. Didelot, A. Spyratou, L.M. Lawson Daku, R. Cerný and H. Hagemann
Inorganic Chemistry, 55 (14) (2016), p7090-7097


DOI:10.1021/acs.inorgchem.6b00931 | unige:85609 | Abstract | Article HTML | Article PDF | Supporting Info

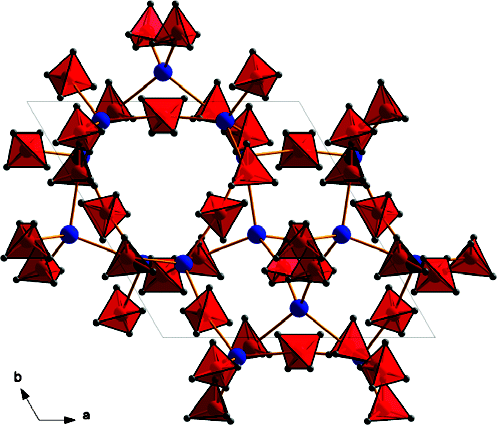
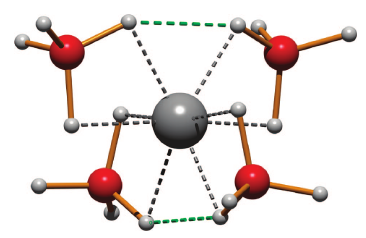
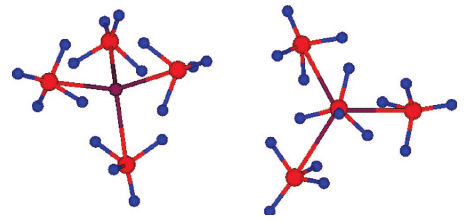
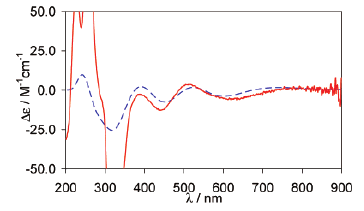
Borohydrides have attained high interest in the past few years due to their high volumetric and gravimetric hydrogen content. Synthesis of di/trimetallic borohydride is a way to alter the thermodynamics of hydrogen release from borohydrides. Previously reported preparations of M(BH4)2 involved chloride containing species such as SrCl2. The presence of residual chloride (or other halide) ions in borohydrides may change their thermodynamic behavior and their decomposition pathway. Pure monometallic borohydrides are needed to study decomposition products without interference from halide impurities. They can also be used as precursors for synthesizing di/trimetallic borohydrides. In this paper we present a way to synthesize halide free alkaline earth metal (Sr, Ba) and europium borohydrides starting with the respective hydrides as precursors. Two novel high temperature polymorphs of Sr and Eu borohydrides and four polymorphs of Ba borohydride have been characterized by synchrotron X-ray powder diffraction, thermal analysis, and Raman and infrared spectroscopy and supported by periodic DFT calculations. The decomposition routes of these borohydrides have also been investigated. In the case of the decomposition of strontium and europium borohydrides, the metal borohydride hydride (M(BH4)H3, M = Sr, Eu) is observed and characterized. Periodic DFT calculations performed on room temperature Ba(BH4)2 revealed the presence of bidentate and tridentate borohydrides.